2022
Orawit Thinnukool
In computer vision, object recognition and image categorization have proven to be difficult challenges. They have, nevertheless, generated responses to a wide range of difficult issues from a variety of fields. Convolution Neural Networks (CNNs) have recently been identified as the most widely proposed deep learning (DL) algorithms in the literature. CNNs have unquestionably delivered cutting-edge achievements, particularly in the areas of image classification, speech recognition, and video processing. However, it has been noticed that the CNN-training assignment demands a large amount of data, which is in low supply, especially in the medical industry, and as a result, the training process takes longer. In this paper, we describe an attention-aware CNN architecture for classifying chest X-ray images to diagnose Pneumonia in order to address the aforementioned difficulties. Attention Modules provide attention-aware properties to the Attention Network. The attention-aware features of various modules alter as the layers become deeper. Using a bottom-up top-down feedforward structure, the feedforward and feedback attention processes are integrated into a single feedforward process inside each attention module.
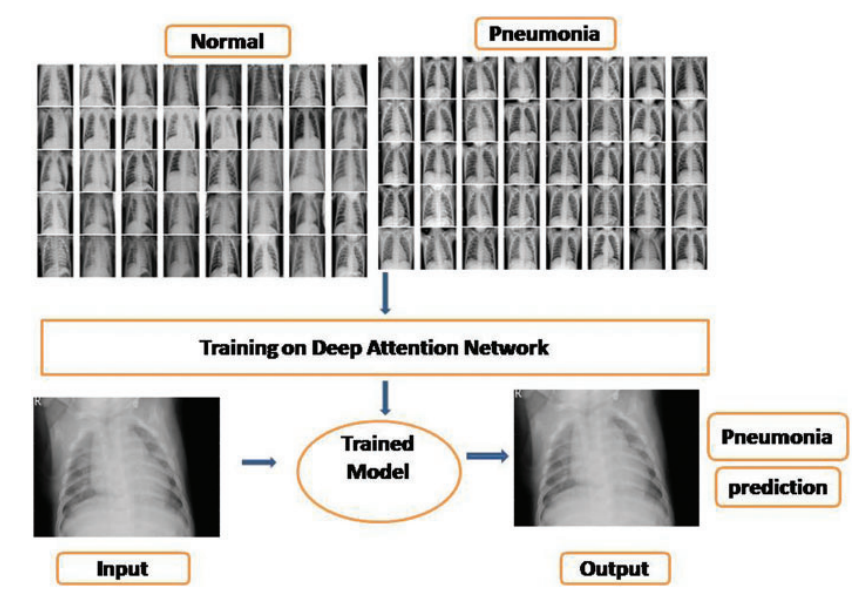
In the present work, a deep neural network (DNN) is combined with an attention mechanism to test the prediction of Pneumonia disease using chest X-ray pictures. To produce attention-aware features, the suggested network was built by merging channel and spatial attention modules in DNN architecture. With this network, we worked on a publicly available Kaggle chest X-ray dataset. Extensive testing was carried out to validate the suggested model. In the experimental results, we attained an accuracy of 95.47% and an F- score of 0.92, indicating that the suggested model outperformed against the baseline models.
2022
Phasit Charoenkwan
Progesterone receptors (PRs) are implicated in various cancers since their presence/absence can determine clinical outcomes. The overstimulation of progesterone can facilitate oncogenesis and thus, its modulation through PR inhibition is urgently needed. To address this issue, a novel stacked ensemble learning approach (termed StackPR) is presented for fast, accurate, and large-scale identification of PR antagonists using only SMILES notation without the need for 3D structural information. We employed six popular machine learning (ML) algorithms (i.e., logistic regression, partial least squares, k-nearest neighbor, support vector machine, extremely randomized trees, and random forest) coupled with twelve conventional molecular descriptors to create 72 baseline models. Then, a genetic algorithm in conjunction with the self-assessment-report approach was utilized to determine m out of the 72 baseline models as means of developing the final meta-predictor using the stacking strategy and tenfold cross-validation test. Experimental results on the independent test dataset show that StackPR achieved impressive predictive performance with an accuracy of 0.966 and Matthew’s coefficient correlation of 0.925. In addition, analysis based on the SHapley Additive exPlanation algorithm and molecular docking indicates that aliphatic hydrocarbons and nitrogen-containing substructures were the most important features for having PR antagonist activity. Finally, we implemented an online webserver using StackPR, which is freely accessible at http://pmlabstack.pythonanywhere.com/StackPR. StackPR is anticipated to be a powerful computational tool for the large-scale identification of unknown PR antagonist candidates for follow-up experimental validation.
https://www.nature.com/articles/s41598-022-20143-5
2022
Phasit Charoenkwan
Amyloid proteins have the ability to form insoluble fibril aggregates that have important pathogenic effects in many tissues. Such amyloidoses are prominently associated with common diseases such as type 2 diabetes, Alzheimer's disease, and Parkinson's disease. There are many types of amyloid proteins, and some proteins that form amyloid aggregates when in a misfolded state. It is difficult to identify such amyloid proteins and their pathogenic properties, but a new and effective approach is by developing effective bioinformatics tools. While several machine learning (ML)-based models for in silico identification of amyloid proteins have been proposed, their predictive performance is limited. In this study, we present AMYPred-FRL, a novel meta-predictor that uses a feature representation learning approach to achieve more accurate amyloid protein identification. AMYPred-FRL combined six well-known ML algorithms (extremely randomized tree, extreme gradient boosting, k-nearest neighbor, logistic regression, random forest, and support vector machine) with ten different sequence-based feature descriptors to generate 60 probabilistic features (PFs), as opposed to state-of-the-art methods developed by a single feature-based approach. A logistic regression recursive feature elimination (LR-RFE) method was used to find the optimal m number of 60 PFs in order to improve the predictive performance. Finally, using the meta-predictor approach, the 20 selected PFs were fed into a logistic regression method to create the final hybrid model (AMYPred-FRL). Both cross-validation and independent tests showed that AMYPred-FRL achieved superior predictive performance than its constituent baseline models. In an extensive independent test, AMYPred-FRL outperformed the existing methods by 5.5% and 16.1%, respectively, with accuracy and MCC of 0.873 and 0.710. To expedite high-throughput prediction, a user-friendly web server of AMYPred-FRL is freely available at http://pmlabstack.pythonanywhere.com/AMYPred-FRL. It is anticipated that AMYPred-FRL will be a useful tool in helping researchers to identify new amyloid proteins.
https://www.nature.com/articles/s41598-022-11897-z
2022
Phasit Charoenkwan
Fast and accurate identification of phage virion proteins (PVPs) would greatly aid facilitation of antibacterial drug discovery and development. Although, several research efforts based on machine learning (ML) methods have been made for in silico identification of PVPs, these methods have certain limitations. Therefore, in this study, we propose a new computational approach, termed SCORPION, (StaCking-based Predictior fOR Phage VIrion PrOteiNs), to accurately identify PVPs using only protein primary sequences. Specifically, we explored comprehensive 13 different feature descriptors from different aspects (i.e., compositional information, composition-transition-distribution information, position-specific information and physicochemical properties) with 10 popular ML algorithms to construct a pool of optimal baseline models. These optimal baseline models were then used to generate probabilistic features (PFs) and considered as a new feature vector. Finally, we utilized a two-step feature selection strategy to determine the optimal PF feature vector and used this feature vector to develop a stacked model (SCORPION). Both tenfold cross-validation and independent test results indicate that SCORPION achieves superior predictive performance than its constitute baseline models and existing methods. We anticipate SCORPION will serve as a useful tool for the cost-effective and large-scale screening of new PVPs. The source codes and datasets for this work are available for downloading in the GitHub repository (https://github.com/saeed344/SCORPION).
https://www.nature.com/articles/s41598-022-08173-5
2022
Phasit Charoenkwan
Antimalarial peptides (AMAPs) varying in length, amino acid composition, charge, conformational structure, hydrophobicity, and amphipathicity reflect their diversity in antimalarial mechanisms. Due to the worldwide major health problem concerning antimicrobial resistance, these peptides possess great therapeutic value owing to their low incidences of drug resistance as compared to conventional antibiotics. Although well-known experimental methods are able to precisely determine the antimalarial activity of peptides, these methods are still time-consuming and costly. Thus, machine learning (ML)-based methods that are capable of identifying AMAPs rapidly by using only sequence information would be beneficial for the high-throughput identification of AMAPs. In this study, we propose the first computational model (termed iAMAP-SCM) for the large-scale identification and characterization of peptides with antimalarial activity by using only sequence information. Specifically, we employed an interpretable scoring card method (SCM) to develop iAMAP-SCM and estimate propensities of 20 amino acids and 400 dipeptides to be AMAPs in a supervised manner. Experimental results showed that iAMAP-SCM could achieve a maximum accuracy and Matthew's coefficient correlation of 0.957 and 0.834, respectively, on the independent test dataset. In addition, SCM-derived propensities of 20 amino acids and selected physicochemical properties were used to provide an understanding of the functional mechanisms of AMAPs. Finally, a user-friendly online computational platform of iAMAP-SCM is publicly available at http://pmlabstack.pythonanywhere.com/iAMAP-SCM. The iAMAP-SCM predictor is anticipated to assist experimental scientists in the high-throughput identification of potential AMAP candidates for the treatment of malaria and other clinical applications.
https://pubs.acs.org/doi/10.1021/acsomega.2c04465
2022
Phasit Charoenkwan
Objectives: To develop a machine learning (ML)-based framework using red blood cell (RBC) parameters for the prediction of the α+-thalassemia trait (α+-thal trait) and to compare the diagnostic performance with a conventional method using a single RBC parameter or a combination of RBC parameters. Methods: A retrospective study was conducted on possible couples at risk for fetus with hemoglobin H (Hb H disease). Subjects with molecularly confirmed normal status (not thalassemia), α+-thal trait, and two-allele α-thalassemia mutation were included. Clinical parameters (age and gender) and RBC parameters (Hb, Hct, MCV, MCH, MCHC, RDW, and RBC count) obtained from their antenatal thalassemia screen were retrieved and analyzed using a machine learning (ML)-based framework and a conventional method. The performance of α+-thal trait prediction was evaluated. Results: In total, 594 cases (female/male: 330/264, mean age: 29.7 ± 6.6 years) were included in the analysis. There were 229 normal controls, 160 cases with the α+-thalassemia trait, and 205 cases in the two-allele α-thalassemia mutation category, respectively. The ML-derived model improved the diagnostic performance, giving a sensitivity of 80% and specificity of 81%. The experimental results indicated that DeepThal achieved a better performance compared with other ML-based methods in terms of the independent test dataset, with an accuracy of 80.77%, sensitivity of 70.59%, and the Matthews correlation coefficient (MCC) of 0.608. Of all the red blood cell parameters, MCH < 28.95 pg as a single parameter had the highest performance in predicting the α+-thal trait with the AUC of 0.857 and 95% CI of 0.816–0.899. The combination model derived from the binary logistic regression analysis exhibited improved performance with the AUC of 0.868 and 95% CI of 0.830–0.906, giving a sensitivity of 80.1% and specificity of 75.1%. Conclusions: The performance of DeepThal in terms of the independent test dataset is sufficient to demonstrate that DeepThal is capable of accurately predicting the α+-thal trait. It is anticipated that DeepThal will be a useful tool for the scientific community in the large-scale prediction of the α+-thal trait.
https://pubmed.ncbi.nlm.nih.gov/36362531/
2022
Phasit Charoenkwan
Discovery of potential drugs requires rapid and precise identification of drug targets. Although traditional experimental methodologies can accurately identify drug targets, they are time-consuming and inappropriate for high-throughput screening. Computational approaches based on machine learning (ML) algorithms can expedite the prediction of druggable proteins; however, the performance of the existing computational methods remains unsatisfactory.
This study proposes a computational tool, SPIDER, to enhance the accurate prediction of druggable proteins. SPIDER employs various feature descriptors pertaining to several aspects, including physicochemical properties, compositional information, and composition-transition-distribution information, coupled with well-known ML algorithms to facilitate the construction of the final meta-predictor. The experimental results showed that SPIDER enabled more precise and robust prediction of druggable proteins than the baseline models and current existing methods in terms of the independent test dataset.
https://www.sciencedirect.com/sdfe/reader/pii/S2589004222011555/pdf
2022
Phasit Charoenkwan
Staphylococcus aureus is deemed to be one of the major causes of hospital and community-acquired infections, especially in methicillin-resistant S. aureus (MRSA) strains. Because antimicrobial peptides have captured attention as novel drug candidates due to their rapid and broad-spectrum antimicrobial activity, anti-MRSA peptides have emerged as potential therapeutics for the treatment of bacterial infections. Although experimental approaches can precisely identify anti-MRSA peptides, they are usually cost-ineffective and labor-intensive. Therefore, computational approaches that are able to identify and characterize anti-MRSA peptides by using sequence information are highly desirable. In this study, we present the first computational approach (termed SCMRSA) for identifying and characterizing anti-MRSA peptides by using sequence information without the use of 3D structural information. In SCMRSA, we employed an interpretable scoring card method (SCM) coupled with the estimated propensity scores of 400 dipeptides. Comparative experiments indicated that SCMRSA was more effective and could outperform several machine learning-based classifiers with an accuracy of 0.960 and Matthews correlation coefficient of 0.848 on the independent test data set. In addition, we employed the SCMRSA-derived propensity scores to provide a more in-depth explanation regarding the functional mechanisms of anti-MRSA peptides. Finally, in order to serve community-wide use of the proposed SCMRSA, we established a user-friendly webserver which can be accessed online at http://pmlabstack.pythonanywhere.com/SCMRSA. SCMRSA is anticipated to be an open-source and useful tool for screening and identifying novel anti-MRSA peptides for follow-up experimental studies.
2022
Phasit Charoenkwan
Tumor homing peptides (THPs) play a crucial role in recognizing and specifically binding to cancer cells. Although experimental approaches can facilitate the precise identification of THPs, they are usually time-consuming, labor-intensive, and not cost-effective. However, computational approaches can identify THPs by utilizing sequence information alone, thus highlighting their great potential for large-scale identification of THPs. Herein, we propose NEPTUNE, a novel computational approach for the accurate and large-scale identification of THPs from sequence information. Specifically, we constructed variant baseline models from multiple feature encoding schemes coupled with six popular machine learning algorithms. Subsequently, we comprehensively assessed and investigated the effects of these baseline models on THP prediction. Finally, the probabilistic information generated by the optimal baseline models is fed into a support vector machine-based classifier to construct the final meta-predictor (NEPTUNE). Cross-validation and independent tests demonstrated that NEPTUNE achieved superior performance for THP prediction compared with its constituent baseline models and the existing methods. Moreover, we employed the powerful SHapley additive exPlanations method to improve the interpretation of NEPTUNE and elucidate the most important features for identifying THPs. Finally, we implemented an online web server using NEPTUNE, which is available at http://pmlabstack.pythonanywhere.com/NEPTUNE. NEPTUNE could be beneficial for the large-scale identification of unknown THP candidates for follow-up experimental validation.
https://www.sciencedirect.com/science/article/abs/pii/S0010482522004826
2022
Phasit Charoenkwan
The development of efficient and effective bioinformatics tools and pipelines for identifying peptides with dipeptidyl peptidase IV (DPP-IV) inhibitory activities from large-scale protein datasets is of great importance for the discovery and development of potential and promising antidiabetic drugs. In this study, we present a novel stacking-based ensemble learning predictor (termed StackDPPIV) designed for identification of DPP-IV inhibitory peptides. Unlike the existing method, which is based on single-feature-based methods, we combined five popular machine learning algorithms in conjunction with ten different feature encodings from multiple perspectives to generate a pool of various baseline models. Subsequently, the probabilistic features derived from these baseline models were systematically integrated and deemed as new feature representations. Finally, in order to improve the predictive performance, the genetic algorithm based on the self-assessment-report was utilized to determine a set of informative probabilistic features and then used the optimal one for developing the final meta-predictor (StackDPPIV). Experiment results demonstrated that StackDPPIV could outperform its constituent baseline models on both the training and independent datasets. Furthermore, StackDPPIV achieved an accuracy of 0.891, MCC of 0.784 and AUC of 0.961, which were 9.4%, 19.0% and 11.4%, respectively, higher than that of the existing method on the independent test. Feature analysis demonstrated that our feature representations had more discriminative ability as compared to conventional feature descriptors, which highlights the combination of different features was essential for the performance improvement. In order to implement the proposed predictor, we had built a user-friendly online web server at http://pmlabstack.pythonanywhere.com/StackDPPIV.
https://www.sciencedirect.com/science/article/abs/pii/S1046202321002711
2021
Orawit Thinnukool
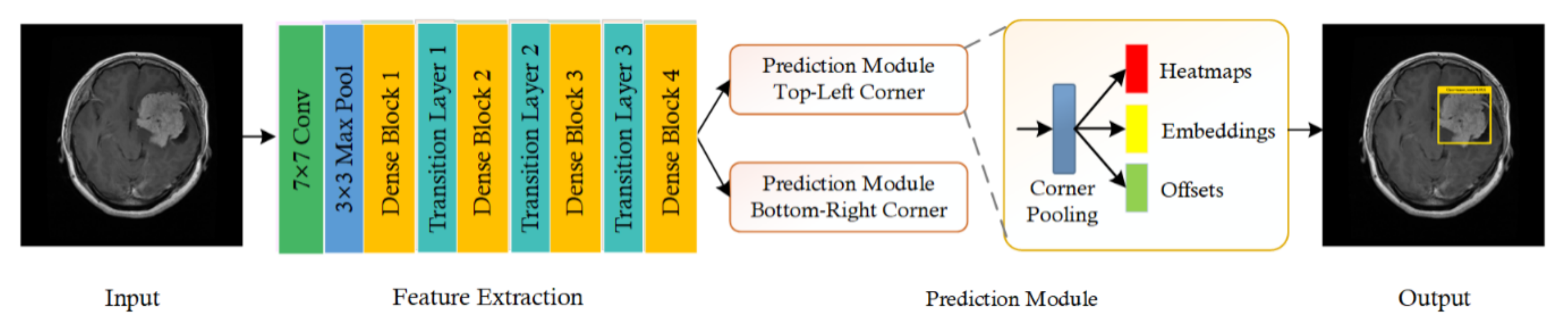
The brain tumor is a deadly disease that is caused by the abnormal growth of brain cells, which affects the human blood cells and nerves. Timely and precise detection of brain tumors is an important task to avoid complex and painful treatment procedures, as it can assist doctors in surgical planning. Manual brain tumor detection is a time-consuming activity and highly dependent on the availability of area experts.
Therefore, it is a need of the hour to design accurate automated systems for the detection and classification of various types of brain tumors. However, the exact localization and categorization of brain tumors is a challenging job due to extensive variations in their size, position, and structure. To deal with the challenges, we have presented a novel approach, namely, DenseNet-41-based CornerNet framework.
The proposed solution comprises three steps. Initially, we develop annotations to locate the exact region of interest. In the second step, a custom CornerNet with DenseNet-41 as a base network is introduced to extract the deep features from the suspected samples. In the last step, the one-stage detector CornerNet is employed to locate and classify several brain tumors. To evaluate the proposed method, we have utilized two databases, namely, the Figshare and Brain MRI datasets, and attained an average accuracy of 98.8% and 98.5%, respectively. Both qualitative and quantitative analysis show that our approach is more proficient and consistent with detecting and classifying various types of brain tumors than other latest technique .
2021
Orawit Thinnukool
we propose a novel computerised approach in this work using deep learning and featuring an ant colony optimisation (ACO) based selection. The proposed method consists of four fundamental steps: data augmentation to solve the imbalanced dataset, fine-tuned pre-trained deep learning models (NasNet Mobile and MobileNet-V2), the fusion of extracted deep features using matrix length, and finally, a selection of the best features using a hybrid ACO and a Neighbourhood Component Analysis (NCA). The best-selected features were eventually passed to many classifiers for final recognition. The experimental process involved an augmented dataset and achieved an average accuracy of 99.7%. Comparison with existing techniques showed that the proposed method was effective.
2021
Orawit Thinnukool
In this study, futuristic and emerging innovations are analyzed, improving COVID-19 effects for the general public. Large data sets need to be advanced so that extensive models related to deep analysis can be used to combat Coronavirus infection, which can be done by applying Artificial intelligence techniques such as Natural Language Processing (NLP), Machine Learning (ML), and Computer vision to varying processing files. This article aims to furnish variation sets of innovations that can be utilized to eliminate COVID-19 and serve as a resource for the coming generations. At last, elaboration associated with future state-of-the-art technologies and the attainable sectors of AI methodologies has been mentioned concerning the post-COVID-19 world to enable the different ideas for dealing with the pandemic-based difficulties.
2021
Pradorn Sureephong
Water, an essential resource for crop production, is becoming increasingly scarce, while cropland continues to expand due to the world’s population growth. Proper irrigation scheduling has been shown to help farmers improve crop yield and quality, resulting in more sustainable water consumption. Soil Moisture (SM), which indicates the amount of water in the soil, is one of the most important crop irrigation parameters. In terms of water usage optimization and crop yield, estimating future soil moisture (forecasting) is an essentially valuable task for crop irrigation. As a result, farmers can base crop irrigation decisions on this parameter. Sensors can be used to estimate this value in real time, which may assist farmers in deciding whether or not to irrigate. The soil moisture value provided by the sensors, on the other hand, is instantaneous and cannot be used to directly compute irrigation parameters such as the best timing or the required water quantity to irrigate. The soil moisture value can, in fact, vary greatly depending on factors such as humidity, weather, and time. Using machine learning methods, these parameters can be used to predict soil moisture levels in the near future. This paper proposes a new Long-Short Term Memory (LSTM)-based model to forecast soil moisture values in the future based on parameters collected from various sensors as a potential solution. To train and validate this model, a real-world dataset containing a set of parameters related to weather forecasting, soil moisture, and other related parameters was collected using smart sensors installed in a greenhouse in Chiang Mai province, Thailand. Preliminary results show that our LSTM-based model performs well in predicting soil moisture with a 0.72% RMSE error and a 0.52% cross-validation error (LSTM), and our Bi-LSTM model with a 0.76% RMSE error and a 0.57% cross-validation error. In the future, we aim to test and validate this model on other similar datasets.